Disclaimer:
Information or materials on this website are not intended to treat or replace your doctor’s advice nor do we endorse any particular product, technique, or idea. It is highly recommended that patients contact their haematologists or health care provider for adequate care and treatment.
Information or materials on this website are not intended to treat or replace your doctor’s advice nor do we endorse any particular product, technique, or idea. It is highly recommended that patients contact their haematologists or health care provider for adequate care and treatment.
What is Sickle Cell?
Definition from the National Heart, Lung and Blood Institute:
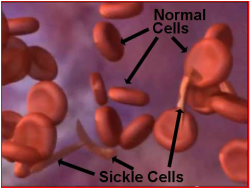
Sickle cell anemia (uh-NEE-me-uh) is the most common form of sickle cell disease (SCD). SCD is a serious disorder in which the body makes sickle-shaped red blood cells. “Sickle-shaped” means that the red blood cells are shaped like a crescent.
Normal red blood cells are disc-shaped and look like doughnuts without holes in the center. They move easily through your blood vessels. Red blood cells contain an iron-rich protein called hemoglobin (HEE-muh-glow-bin). This protein carries oxygen from the lungs to the rest of the body.
Sickle cells contain abnormal hemoglobin called sickle hemoglobin or hemoglobin S. Sickle hemoglobin causes the cells to develop a sickle, or crescent, shape.
Sickle cells are stiff and sticky. They tend to block blood flow in the blood vessels of the limbs and organs. Blocked blood flow can cause pain and organ damage. It can also raise the risk for infection.
Definition from the National Heart, Lung and Blood Institute:
Sickle cell anemia (uh-NEE-me-uh) is the most common form of sickle cell disease (SCD). SCD is a serious disorder in which the body makes sickle-shaped red blood cells. “Sickle-shaped” means that the red blood cells are shaped like a crescent.
Normal red blood cells are disc-shaped and look like doughnuts without holes in the center. They move easily through your blood vessels. Red blood cells contain an iron-rich protein called hemoglobin (HEE-muh-glow-bin). This protein carries oxygen from the lungs to the rest of the body.
Sickle cells contain abnormal hemoglobin called sickle hemoglobin or hemoglobin S. Sickle hemoglobin causes the cells to develop a sickle, or crescent, shape.
Sickle cells are stiff and sticky. They tend to block blood flow in the blood vessels of the limbs and organs. Blocked blood flow can cause pain and organ damage. It can also raise the risk for infection.
How do you get Sickle Cell?
Definition from Medicinenet.com
How is sickle cell anemia inherited?
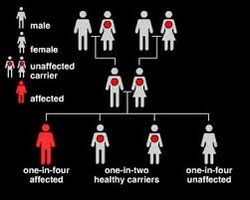
Sickle cell anemia is inherited as an autosomal (meaning that the gene is not linked to a sex chromosome) recessive condition whereas sickle cell trait is inherited as an autosomal dominant trait. This means that the gene can be passed on from a parent carrying it to male and female children. In order for sickle cell anemia to occur, a sickle cell gene must be inherited from both the mother and the father, so that the child has two sickle cell genes.
The inheritance of just one sickle gene is called sickle cell trait or the "carrier" state. Sickle cell trait does not cause sickle cell anemia. Persons with sickle cell trait usually do not have many symptoms of disease and have normal hospitalization rates and life expectancies. Sickle cell trait is present in some two million blacks in the United States (8% of the U.S. black population at birth). When two carriers of sickle cell trait mate, their offspring have a one in four chance of having sickle cell anemia. (In some parts of Africa, one in five persons is a carrier for sickle cell trait.)
Link:
Sickle Cell Anemia genetic description - video:
Written by Paulo Cesar Naoum, BSc, Phd and Alia F. Naoum , MtSc. MSc
http://www.youtube.com/watch?v=R4-c3hUhhyc
Definition from Medicinenet.com
How is sickle cell anemia inherited?
Sickle cell anemia is inherited as an autosomal (meaning that the gene is not linked to a sex chromosome) recessive condition whereas sickle cell trait is inherited as an autosomal dominant trait. This means that the gene can be passed on from a parent carrying it to male and female children. In order for sickle cell anemia to occur, a sickle cell gene must be inherited from both the mother and the father, so that the child has two sickle cell genes.
The inheritance of just one sickle gene is called sickle cell trait or the "carrier" state. Sickle cell trait does not cause sickle cell anemia. Persons with sickle cell trait usually do not have many symptoms of disease and have normal hospitalization rates and life expectancies. Sickle cell trait is present in some two million blacks in the United States (8% of the U.S. black population at birth). When two carriers of sickle cell trait mate, their offspring have a one in four chance of having sickle cell anemia. (In some parts of Africa, one in five persons is a carrier for sickle cell trait.)
Link:
Sickle Cell Anemia genetic description - video:
Written by Paulo Cesar Naoum, BSc, Phd and Alia F. Naoum , MtSc. MSc
http://www.youtube.com/watch?v=R4-c3hUhhyc
Normal Red Blood Cells and Sickle Cells
Definition from the National Heart, Lung and Blood Institute:
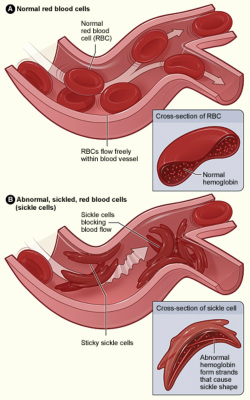
Figure A shows normal red blood cells flowing freely in a blood vessel. The inset image shows a cross-section of a normal red blood cell with normal hemoglobin. Figure B shows abnormal, sickled red blood cells blocking blood flow in a blood vessel. The inset image shows a cross-section of a sickle cell with abnormal (sickle) hemoglobin forming abnormal strands.
Overview
Sickle cell anemia is one type of anemia. Anemia is a condition in which your blood has a lower than normal number of red blood cells. This condition also can occur if your red blood cells don't contain enough hemoglobin.
Red blood cells are made in the spongy marrow inside the larger bones of the body. Bone marrow is always making new red blood cells to replace old ones. Normal red blood cells live about 120 days in the bloodstream and then die. They carry oxygen and remove carbon dioxide (a waste product) from your body.
In sickle cell anemia, the abnormal sickle cells usually die after only about 10 to 20 days. The bone marrow can't make new red blood cells fast enough to replace the dying ones.
Sickle cell anemia is an inherited, lifelong disease. People who have the disease are born with it. They inherit two genes for sickle hemoglobin—one from each parent.
People who inherit a sickle hemoglobin gene from one parent and a normal gene from the other parent have a condition called sickle cell trait.
Sickle cell trait is different than sickle cell anemia. People who have sickle cell trait don't have the disease. Like people who have sickle cell anemia, people who have sickle cell trait can pass the sickle hemoglobin gene to their children.
Outlook
Sickle cell anemia has no widely available cure. However, treatments to improve the anemia and lower complications can help with the symptoms and complications of the disease in both children and adults. Blood and marrow stem cell transplants may offer a cure for a small number of people.
Over the past 100 years, doctors have learned a great deal about sickle cell anemia. They know its causes, how it affects the body, and how to treat many of its complications.
Sickle cell anemia varies from person to person. Some people who have the disease have chronic (long-term) pain or fatigue (tiredness). However, with proper care and treatment, many people who have the disease can have improved quality of life and reasonable health much of the time.
Because of improved treatments and care, people who have sickle cell anemia are now living into their forties or fifties, or longer.
Other Names for Sickle Cell Anemia
Medical links:
Mayo Clinic
http://www.mayoclinic.com/health/sickle-cell-anemia/DS00324
University of Illinois - Hospital and Health Sciences System
http://hospital.uillinois.edu/Patient_Care_Services/Sickle_Cell/Managing_Sickle_Cell.html
WebMD - Sickle Cell Disease
http://www.webmd.com/a-to-z-guides/sickle-cell-disease-topic-overview
Definition from the National Heart, Lung and Blood Institute:
Figure A shows normal red blood cells flowing freely in a blood vessel. The inset image shows a cross-section of a normal red blood cell with normal hemoglobin. Figure B shows abnormal, sickled red blood cells blocking blood flow in a blood vessel. The inset image shows a cross-section of a sickle cell with abnormal (sickle) hemoglobin forming abnormal strands.
Overview
Sickle cell anemia is one type of anemia. Anemia is a condition in which your blood has a lower than normal number of red blood cells. This condition also can occur if your red blood cells don't contain enough hemoglobin.
Red blood cells are made in the spongy marrow inside the larger bones of the body. Bone marrow is always making new red blood cells to replace old ones. Normal red blood cells live about 120 days in the bloodstream and then die. They carry oxygen and remove carbon dioxide (a waste product) from your body.
In sickle cell anemia, the abnormal sickle cells usually die after only about 10 to 20 days. The bone marrow can't make new red blood cells fast enough to replace the dying ones.
Sickle cell anemia is an inherited, lifelong disease. People who have the disease are born with it. They inherit two genes for sickle hemoglobin—one from each parent.
People who inherit a sickle hemoglobin gene from one parent and a normal gene from the other parent have a condition called sickle cell trait.
Sickle cell trait is different than sickle cell anemia. People who have sickle cell trait don't have the disease. Like people who have sickle cell anemia, people who have sickle cell trait can pass the sickle hemoglobin gene to their children.
Outlook
Sickle cell anemia has no widely available cure. However, treatments to improve the anemia and lower complications can help with the symptoms and complications of the disease in both children and adults. Blood and marrow stem cell transplants may offer a cure for a small number of people.
Over the past 100 years, doctors have learned a great deal about sickle cell anemia. They know its causes, how it affects the body, and how to treat many of its complications.
Sickle cell anemia varies from person to person. Some people who have the disease have chronic (long-term) pain or fatigue (tiredness). However, with proper care and treatment, many people who have the disease can have improved quality of life and reasonable health much of the time.
Because of improved treatments and care, people who have sickle cell anemia are now living into their forties or fifties, or longer.
Other Names for Sickle Cell Anemia
- HbS disease
- Hemoglobin S disease
- Hemoglobin SS disease
- Sickle cell disease (a broad term that includes sickle cell anemia)
- Sickle cell disorders (a broad group of conditions that includes sickle cell anemia)
- Sickling disorder due to hemoglobin S
Medical links:
Mayo Clinic
http://www.mayoclinic.com/health/sickle-cell-anemia/DS00324
University of Illinois - Hospital and Health Sciences System
http://hospital.uillinois.edu/Patient_Care_Services/Sickle_Cell/Managing_Sickle_Cell.html
WebMD - Sickle Cell Disease
http://www.webmd.com/a-to-z-guides/sickle-cell-disease-topic-overview
What are the symptoms?
Definition from the National Heart, Lung and Blood Institute:
The signs and symptoms of sickle cell anemia vary. Some people have mild symptoms. Others have very severe symptoms and often are hospitalized for treatment.
Sickle cell anemia is present at birth, but many infants don't show any signs until after 4 months of age.
The most common signs and symptoms are linked to anemia and pain. Other signs and symptoms are linked to the disease's complications.
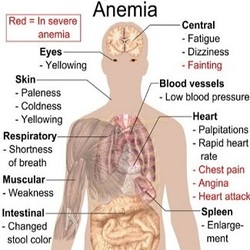
Signs and Symptoms Related to Anemia
The most common symptom of anemia is fatigue (feeling tired or weak). Other signs and symptoms of anemia include:
Sudden pain throughout the body is a common symptom of sickle cell anemia. This pain is called a sickle cell crisis. Sickle cell crises often affect the bones, lungs, abdomen, and joints.
These crises occur when sickled red blood cells block blood flow to the limbs and organs. This can cause pain and organ damage.
The pain from sickle cell anemia can be acute or chronic, but acute pain is more common. Acute pain is sudden and can range from mild to very severe. The pain usually lasts from hours to as long as a week or more.
Many people who have sickle cell anemia also have chronic pain, especially in their bones. Chronic pain often lasts for weeks or months and can be hard to bear and mentally draining. Chronic pain may limit your daily activities.
Almost all people who have sickle cell anemia have painful crises at some point in their lives. Some have these crises less than once a year. Others may have crises once a month or more. Repeated crises can damage the bones, kidneys, lungs, eyes, heart, and liver. This type of damage happens more often in adults than in children.
Many factors can play a role in sickle cell crises. Often, more than one factor is involved and the exact cause isn't known.
You can control some factors. For example, the risk of a sickle cell crisis increases if you're dehydrated (your body doesn't have enough fluids). Drinking plenty of fluids can lower the risk of a painful crisis.
You can't control other factors, such as infections.
Painful crises are the leading cause of emergency room visits and hospital stays for people who have sickle cell anemia.
Complications of Sickle Cell Anemia
Sickle cell crises can affect many parts of the body and cause many complications.
Hand-Foot Syndrome
Sickle cells can block the small blood vessels in the hands and feet in children (usually those younger than 4 years of age). This condition is called hand-foot syndrome. It can lead to pain, swelling, and fever.
Swelling often occurs on the back of the hands and feet and moves into the fingers and toes. One or both hands and/or feet might be affected at the same time.
Splenic Crisis
The spleen is an organ in the abdomen. Normally, it filters out abnormal red blood cells and helps fight infections. Sometimes the spleen may trap red blood cells that should be in the bloodstream. This causes the spleen to grow large and leads to anemia.
If the spleen traps too many red blood cells, you may need blood transfusions until your body can make more cells and recover.
Infections
Both children and adults who have sickle cell anemia may get infections easily and have a hard time fighting them. This is because sickle cell anemia can damage the spleen, an organ that helps fight infections.
Infants and young children who have damaged spleens are more likely to get serious infections that can kill them within hours or days. Bloodstream infections are the most common cause of death in young children who have sickle cell anemia.
Medicines and vaccines can help prevent severe illness and death. For example, vaccines are available for infections such as meningitis, influenza, and hepatitis.
Getting treatment right away for high fevers (which can be a sign of a severe infection) also helps prevent death in infants and children who have sickle cell anemia.
It's also important to get treatment right away for a cough, problems breathing, bone pain, and headaches.
Acute Chest Syndrome
Acute chest syndrome is a life-threatening condition linked to sickle cell anemia. This syndrome is similar to pneumonia. An infection or sickle cells trapped in the lungs can cause acute chest syndrome.
People who have this condition often have chest pain, shortness of breath, and fever. They also often have low oxygen levels and abnormal chest x ray results.
Pulmonary Hypertension
Damage to the small blood vessels in the lungs makes it hard for the heart to pump blood through the lungs. This causes blood pressure in the lungs to rise.
Increased blood pressure in the lungs is called pulmonary hypertension (PH). Shortness of breath and fatigue are the main symptoms of PH.
Delayed Growth and Puberty in Children
Children who have sickle cell anemia often grow more slowly than other children. They may reach puberty later. A shortage of red blood cells causes the slow growth rate. Adults who have sickle cell anemia often are slender or smaller in size than other adults.
Stroke
Two forms of stroke can occur in people who have sickle cell anemia. One form occurs if a blood vessel in the brain is damaged and blocked. This type of stroke occurs more often in children than adults. The other form of stroke occurs if a blood vessel in the brain bursts.
Either type of stroke can cause learning problems and lasting brain damage, long-term disability, paralysis (an inability to move), or death.
Eye Problems
Sickle cells also can affect the small blood vessels that deliver oxygen-rich blood to the eyes. Sickle cells can block these vessels or cause them to break open and bleed. This can damage the retinas—thin layers of tissue at the back of the eyes. The retinas take the images you see and send them to your brain.
This damage can cause serious problems, including blindness.
Priapism
Males who have sickle cell anemia may have painful, unwanted erections. This condition is called priapism (PRI-a-pizm). It happens because the sickle cells block blood flow out of an erect penis. Over time, priapism can damage the penis and lead to impotence.
Gallstones
When red blood cells die, they release their hemoglobin. The body breaks down this protein into a compound called bilirubin. Too much bilirubin in the body can cause stones to form in the gallbladder, called gallstones.
Gallstones may cause steady pain that lasts for 30 minutes or more in the upper right side of the belly, under the right shoulder, or between the shoulder blades. The pain may happen after eating fatty meals.
People who have gallstones may have nausea (feeling sick to the stomach), vomiting, fever, sweating, chills, clay-colored stools, or jaundice.
Ulcers on the Legs
Sickle cell ulcers (sores) usually begin as small, raised, crusted sores on the lower third of the leg. Leg sores may occur more often in males than in females. These sores usually develop in people who are aged 10 years or older.
The cause of sickle cell ulcers isn't clear. The number of ulcers can vary from one to many. Some heal quickly, but others persist for years or come back after healing.
Multiple Organ Failure
Multiple organ failure is rare, but serious. It happens if you have a sickle cell crisis that causes two out of three major organs (lungs, liver, or kidneys) to fail. Often, multiple organ failure occurs during an unusually severe pain crisis.
Symptoms of this complication are fever, rapid heartbeat, problems breathing, and changes in mental status (such as sudden tiredness or confusion).
A simple blood test, done at any time during a person's lifespan, can detect whether he or she has sickle hemoglobin. However, early diagnosis is very important.
In the United States, all States mandate testing for sickle cell anemia as part of their newborn screening programs. The test uses blood from the same blood samples used for other routine newborn screening tests. The test can show whether a newborn infant has sickle hemoglobin.
Test results are sent to the doctor who ordered the test and to the baby's primary care doctor. It's important to give the correct contact information to the hospital. This allows the baby's doctor to get the test results as quickly as possible.
Health providers from a newborn screening followup program may contact you directly to make sure you're aware of the test results.
If the test shows some sickle hemoglobin, a second blood test is done to confirm the diagnosis. The second test should be done as soon as possible and within the first few months of life.
The primary care doctor may send you to a hematologist for a second blood test. A hematologist is a doctor who specializes in blood diseases and disorders. This doctor also can provide treatment for sickle cell disease if needed.
Doctors also can diagnose sickle cell disease before birth. This is done using a sample of amniotic fluid or tissue taken from the placenta. (Amniotic fluid is the fluid in the sac surrounding a growing embryo. The placenta is the organ that attaches the umbilical cord to the mother's womb.)
Testing before birth can be done as early as 10 weeks into the pregnancy. This testing looks for the sickle hemoglobin gene, rather than the abnormal hemoglobin that the gene makes.
Links
WebMd - SC Symptoms
http://www.webmd.com/a-to-z-guides/sickle-cell-disease-symptoms
WebMD - What Happens
http://www.webmd.com/a-to-z-guides/sickle-cell-disease-what-happens
Definition from the National Heart, Lung and Blood Institute:
The signs and symptoms of sickle cell anemia vary. Some people have mild symptoms. Others have very severe symptoms and often are hospitalized for treatment.
Sickle cell anemia is present at birth, but many infants don't show any signs until after 4 months of age.
The most common signs and symptoms are linked to anemia and pain. Other signs and symptoms are linked to the disease's complications.
Signs and Symptoms Related to Anemia
The most common symptom of anemia is fatigue (feeling tired or weak). Other signs and symptoms of anemia include:
- Shortness of breath
- Dizziness
- Headaches
- Coldness in the hands and feet
- Paler than normal skin or mucous membranes (the tissue that lines your nose, mouth, and other organs and body cavities)
- Jaundice (a yellowish color of the skin or whites of the eyes)
Sudden pain throughout the body is a common symptom of sickle cell anemia. This pain is called a sickle cell crisis. Sickle cell crises often affect the bones, lungs, abdomen, and joints.
These crises occur when sickled red blood cells block blood flow to the limbs and organs. This can cause pain and organ damage.
The pain from sickle cell anemia can be acute or chronic, but acute pain is more common. Acute pain is sudden and can range from mild to very severe. The pain usually lasts from hours to as long as a week or more.
Many people who have sickle cell anemia also have chronic pain, especially in their bones. Chronic pain often lasts for weeks or months and can be hard to bear and mentally draining. Chronic pain may limit your daily activities.
Almost all people who have sickle cell anemia have painful crises at some point in their lives. Some have these crises less than once a year. Others may have crises once a month or more. Repeated crises can damage the bones, kidneys, lungs, eyes, heart, and liver. This type of damage happens more often in adults than in children.
Many factors can play a role in sickle cell crises. Often, more than one factor is involved and the exact cause isn't known.
You can control some factors. For example, the risk of a sickle cell crisis increases if you're dehydrated (your body doesn't have enough fluids). Drinking plenty of fluids can lower the risk of a painful crisis.
You can't control other factors, such as infections.
Painful crises are the leading cause of emergency room visits and hospital stays for people who have sickle cell anemia.
Complications of Sickle Cell Anemia
Sickle cell crises can affect many parts of the body and cause many complications.
Hand-Foot Syndrome
Sickle cells can block the small blood vessels in the hands and feet in children (usually those younger than 4 years of age). This condition is called hand-foot syndrome. It can lead to pain, swelling, and fever.
Swelling often occurs on the back of the hands and feet and moves into the fingers and toes. One or both hands and/or feet might be affected at the same time.
Splenic Crisis
The spleen is an organ in the abdomen. Normally, it filters out abnormal red blood cells and helps fight infections. Sometimes the spleen may trap red blood cells that should be in the bloodstream. This causes the spleen to grow large and leads to anemia.
If the spleen traps too many red blood cells, you may need blood transfusions until your body can make more cells and recover.
Infections
Both children and adults who have sickle cell anemia may get infections easily and have a hard time fighting them. This is because sickle cell anemia can damage the spleen, an organ that helps fight infections.
Infants and young children who have damaged spleens are more likely to get serious infections that can kill them within hours or days. Bloodstream infections are the most common cause of death in young children who have sickle cell anemia.
Medicines and vaccines can help prevent severe illness and death. For example, vaccines are available for infections such as meningitis, influenza, and hepatitis.
Getting treatment right away for high fevers (which can be a sign of a severe infection) also helps prevent death in infants and children who have sickle cell anemia.
It's also important to get treatment right away for a cough, problems breathing, bone pain, and headaches.
Acute Chest Syndrome
Acute chest syndrome is a life-threatening condition linked to sickle cell anemia. This syndrome is similar to pneumonia. An infection or sickle cells trapped in the lungs can cause acute chest syndrome.
People who have this condition often have chest pain, shortness of breath, and fever. They also often have low oxygen levels and abnormal chest x ray results.
Pulmonary Hypertension
Damage to the small blood vessels in the lungs makes it hard for the heart to pump blood through the lungs. This causes blood pressure in the lungs to rise.
Increased blood pressure in the lungs is called pulmonary hypertension (PH). Shortness of breath and fatigue are the main symptoms of PH.
Delayed Growth and Puberty in Children
Children who have sickle cell anemia often grow more slowly than other children. They may reach puberty later. A shortage of red blood cells causes the slow growth rate. Adults who have sickle cell anemia often are slender or smaller in size than other adults.
Stroke
Two forms of stroke can occur in people who have sickle cell anemia. One form occurs if a blood vessel in the brain is damaged and blocked. This type of stroke occurs more often in children than adults. The other form of stroke occurs if a blood vessel in the brain bursts.
Either type of stroke can cause learning problems and lasting brain damage, long-term disability, paralysis (an inability to move), or death.
Eye Problems
Sickle cells also can affect the small blood vessels that deliver oxygen-rich blood to the eyes. Sickle cells can block these vessels or cause them to break open and bleed. This can damage the retinas—thin layers of tissue at the back of the eyes. The retinas take the images you see and send them to your brain.
This damage can cause serious problems, including blindness.
Priapism
Males who have sickle cell anemia may have painful, unwanted erections. This condition is called priapism (PRI-a-pizm). It happens because the sickle cells block blood flow out of an erect penis. Over time, priapism can damage the penis and lead to impotence.
Gallstones
When red blood cells die, they release their hemoglobin. The body breaks down this protein into a compound called bilirubin. Too much bilirubin in the body can cause stones to form in the gallbladder, called gallstones.
Gallstones may cause steady pain that lasts for 30 minutes or more in the upper right side of the belly, under the right shoulder, or between the shoulder blades. The pain may happen after eating fatty meals.
People who have gallstones may have nausea (feeling sick to the stomach), vomiting, fever, sweating, chills, clay-colored stools, or jaundice.
Ulcers on the Legs
Sickle cell ulcers (sores) usually begin as small, raised, crusted sores on the lower third of the leg. Leg sores may occur more often in males than in females. These sores usually develop in people who are aged 10 years or older.
The cause of sickle cell ulcers isn't clear. The number of ulcers can vary from one to many. Some heal quickly, but others persist for years or come back after healing.
Multiple Organ Failure
Multiple organ failure is rare, but serious. It happens if you have a sickle cell crisis that causes two out of three major organs (lungs, liver, or kidneys) to fail. Often, multiple organ failure occurs during an unusually severe pain crisis.
Symptoms of this complication are fever, rapid heartbeat, problems breathing, and changes in mental status (such as sudden tiredness or confusion).
A simple blood test, done at any time during a person's lifespan, can detect whether he or she has sickle hemoglobin. However, early diagnosis is very important.
In the United States, all States mandate testing for sickle cell anemia as part of their newborn screening programs. The test uses blood from the same blood samples used for other routine newborn screening tests. The test can show whether a newborn infant has sickle hemoglobin.
Test results are sent to the doctor who ordered the test and to the baby's primary care doctor. It's important to give the correct contact information to the hospital. This allows the baby's doctor to get the test results as quickly as possible.
Health providers from a newborn screening followup program may contact you directly to make sure you're aware of the test results.
If the test shows some sickle hemoglobin, a second blood test is done to confirm the diagnosis. The second test should be done as soon as possible and within the first few months of life.
The primary care doctor may send you to a hematologist for a second blood test. A hematologist is a doctor who specializes in blood diseases and disorders. This doctor also can provide treatment for sickle cell disease if needed.
Doctors also can diagnose sickle cell disease before birth. This is done using a sample of amniotic fluid or tissue taken from the placenta. (Amniotic fluid is the fluid in the sac surrounding a growing embryo. The placenta is the organ that attaches the umbilical cord to the mother's womb.)
Testing before birth can be done as early as 10 weeks into the pregnancy. This testing looks for the sickle hemoglobin gene, rather than the abnormal hemoglobin that the gene makes.
Links
WebMd - SC Symptoms
http://www.webmd.com/a-to-z-guides/sickle-cell-disease-symptoms
WebMD - What Happens
http://www.webmd.com/a-to-z-guides/sickle-cell-disease-what-happens
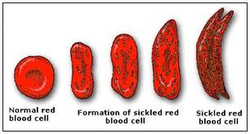
How is Sickle Cell linked to Malaria? How does Sickle Cell protect against severe Malaria?
Article from Science Daily (Note: There are several related links on the Science Daily website. See below):
Mystery Solved: How Sickle Hemoglobin Protects Against Malaria -- Apr. 29, 2011
The latest issue of the journal Cellcarries an article that is likely to help solve one of the long-standing mysteries of biomedicine. In a study that challenges currently held views, researchers at the Instituto Gulbenkian de Ciência (IGC), in Portugal, unravel the molecular mechanism whereby sickle cell hemoglobin confers a survival advantage against malaria, the disease caused by Plasmodium infection. These findings, by the research team lead by Miguel P. Soares, open the way to new therapeutic interventions against malaria, a disease that continues to inflict tremendous medical, social and economic burdens to a large proportion of the human population.
Sickle cell anemia is a blood disease in which red blood cells reveal an abnormal crescent (or sickle) shape when observed under a conventional microscope. It is an inherited disorder -- the first ever to be attributed to a specific genetic modification (mutation), in 1949 by Linus Pauling (two-times Nobel laureate, for Chemistry in 1954, and Peace, in 1962). The cause of sickle cell anemia was attributed unequivocally to a single base substitution in the DNA sequence of the gene encoding the beta chain of hemoglobin, the protein that carries oxygen in red blood cells.
Only those individual that inherit two copies of the sickle mutation (one from their mother and the other from their father) develop sickle cell anemia. If untreated, these individuals have a shorter than normal life expectancy and as such it would be expected that this mutation would be rare in human populations. This is however, far from being the case. Observations made during the mid-20th century and building on Pauling's findings, revealed that the sickle mutation is, in fact, highly, selected in populations from areas of the world were malaria is very frequent, with sometimes 10-40% of the population carrying this mutation.
Individuals carrying just one copy of the sickle mutation (inherited from either the father or mother) were known not to develop sickle cell anemia, leading rather normal lives. However, it was found that these same individuals, said to carry the sickle cell trait, were in fact highly protected against malaria, thus explaining the high prevalence of this mutation in geographical areas where malaria is endemic.
These findings lead to the widespread believe in the medical community that understanding the mechanism whereby sickle cell trait protects against malaria would provide critical insight into developing treatment or a possible cure for this devastating disease, responsible for over a million premature deaths in sub-Saharan Africa. Despite several decades of research, the mechanism underlying this protective effect remained elusive. Until now.
Several studies suggested that, in one way or another, sickle hemoglobin might get in the way of the Plasmodium parasite infecting red blood cells, reducing the number of parasites that actually infect the host and thus conferring some protection against the disease. The IGC team's results challenge this explanation.
In painstakingly detailed work, Ana Ferreira, a post-doctoral researcher in Miguel Soares' laboratory, demonstrated that mice obtained from Prof. Yves Beuzard's laboratory, that had been genetically engineered to produce one copy of sickle hemoglobin similar to sickle cell trait, do not succumb to cerebral malaria, thus reproducing what happens in humans.
When Prof. Ingo Bechman observed the brains of these mice he confirmed that the lesions associated with the development of cerebral malaria where absent, despite the presence of the parasite.
Ana Ferreira went on to show that the protection afforded by sickle hemoglobin in these mice, acts without interfering directly with the parasite's ability to infect the host red blood cells. As Miguel Soares describes it, "sickle hemoglobin makes the host tolerant to the parasite."
Through a series of genetic experiments, Ana Ferreira was able to show that the main player in this protective effect is heme oxygenase-1 (HO-1), an enzyme whose expression is strongly induced by sickle hemoglobin. This enzyme, that produces the gas carbon monoxide, had been previously shown by the laboratory of Miguel Soares to confer protection against cerebral malaria. In the process of dissecting further this mechanism of protection Ana Ferreira demonstrated that when produced in response to sickle hemoglobin the same gas, carbon monoxide, protected the infected host from succumbing to cerebral malaria without interfering with the life cycle of the parasite inside its red blood cells.
Miguel Soares and his team believe that the mechanism they have identified for sickle cell trait may be a general mechanism acting in other red blood cell genetic diseases that are also know to protect against malaria in human populations: "Due to its protective effect against malaria, the sickle mutation may have been naturally selected in sub-Saharan Africa, where malaria is endemic and one of the major causes of death. Similarly, other clinically silent mutations may have been selected throughout evolution, for their ability to provide survival advantage against Plasmodium infection."
This research was carried out the at the IGC in collaboration with the Team of Prof. Yves Beuzard (Université Paris VII et XI, France), an expert in sickle cell anemia, and Prof. Ingo Bechman an expert in neuropathological diseases (Institute of Anatomy, University of Leipzig, Germany). Other IGC researchers involved in this study are Ivo Marguti, Viktória Jeney, Ângelo Chora, Nuno Palha and Sofia Rebelo. This project was funded by Fundação para a Ciência e a Tecnologia (Portugal), GEMI Fund Linde Healthcare and the European Commission's Framework Programme 7.
Related Links / Articles:
Protection from Severe Malaria Explained
Naturally-Occurring Protection Against Severe Malaria Demonstrated
Article from Science Daily (Note: There are several related links on the Science Daily website. See below):
Mystery Solved: How Sickle Hemoglobin Protects Against Malaria -- Apr. 29, 2011
The latest issue of the journal Cellcarries an article that is likely to help solve one of the long-standing mysteries of biomedicine. In a study that challenges currently held views, researchers at the Instituto Gulbenkian de Ciência (IGC), in Portugal, unravel the molecular mechanism whereby sickle cell hemoglobin confers a survival advantage against malaria, the disease caused by Plasmodium infection. These findings, by the research team lead by Miguel P. Soares, open the way to new therapeutic interventions against malaria, a disease that continues to inflict tremendous medical, social and economic burdens to a large proportion of the human population.
Sickle cell anemia is a blood disease in which red blood cells reveal an abnormal crescent (or sickle) shape when observed under a conventional microscope. It is an inherited disorder -- the first ever to be attributed to a specific genetic modification (mutation), in 1949 by Linus Pauling (two-times Nobel laureate, for Chemistry in 1954, and Peace, in 1962). The cause of sickle cell anemia was attributed unequivocally to a single base substitution in the DNA sequence of the gene encoding the beta chain of hemoglobin, the protein that carries oxygen in red blood cells.
Only those individual that inherit two copies of the sickle mutation (one from their mother and the other from their father) develop sickle cell anemia. If untreated, these individuals have a shorter than normal life expectancy and as such it would be expected that this mutation would be rare in human populations. This is however, far from being the case. Observations made during the mid-20th century and building on Pauling's findings, revealed that the sickle mutation is, in fact, highly, selected in populations from areas of the world were malaria is very frequent, with sometimes 10-40% of the population carrying this mutation.
Individuals carrying just one copy of the sickle mutation (inherited from either the father or mother) were known not to develop sickle cell anemia, leading rather normal lives. However, it was found that these same individuals, said to carry the sickle cell trait, were in fact highly protected against malaria, thus explaining the high prevalence of this mutation in geographical areas where malaria is endemic.
These findings lead to the widespread believe in the medical community that understanding the mechanism whereby sickle cell trait protects against malaria would provide critical insight into developing treatment or a possible cure for this devastating disease, responsible for over a million premature deaths in sub-Saharan Africa. Despite several decades of research, the mechanism underlying this protective effect remained elusive. Until now.
Several studies suggested that, in one way or another, sickle hemoglobin might get in the way of the Plasmodium parasite infecting red blood cells, reducing the number of parasites that actually infect the host and thus conferring some protection against the disease. The IGC team's results challenge this explanation.
In painstakingly detailed work, Ana Ferreira, a post-doctoral researcher in Miguel Soares' laboratory, demonstrated that mice obtained from Prof. Yves Beuzard's laboratory, that had been genetically engineered to produce one copy of sickle hemoglobin similar to sickle cell trait, do not succumb to cerebral malaria, thus reproducing what happens in humans.
When Prof. Ingo Bechman observed the brains of these mice he confirmed that the lesions associated with the development of cerebral malaria where absent, despite the presence of the parasite.
Ana Ferreira went on to show that the protection afforded by sickle hemoglobin in these mice, acts without interfering directly with the parasite's ability to infect the host red blood cells. As Miguel Soares describes it, "sickle hemoglobin makes the host tolerant to the parasite."
Through a series of genetic experiments, Ana Ferreira was able to show that the main player in this protective effect is heme oxygenase-1 (HO-1), an enzyme whose expression is strongly induced by sickle hemoglobin. This enzyme, that produces the gas carbon monoxide, had been previously shown by the laboratory of Miguel Soares to confer protection against cerebral malaria. In the process of dissecting further this mechanism of protection Ana Ferreira demonstrated that when produced in response to sickle hemoglobin the same gas, carbon monoxide, protected the infected host from succumbing to cerebral malaria without interfering with the life cycle of the parasite inside its red blood cells.
Miguel Soares and his team believe that the mechanism they have identified for sickle cell trait may be a general mechanism acting in other red blood cell genetic diseases that are also know to protect against malaria in human populations: "Due to its protective effect against malaria, the sickle mutation may have been naturally selected in sub-Saharan Africa, where malaria is endemic and one of the major causes of death. Similarly, other clinically silent mutations may have been selected throughout evolution, for their ability to provide survival advantage against Plasmodium infection."
This research was carried out the at the IGC in collaboration with the Team of Prof. Yves Beuzard (Université Paris VII et XI, France), an expert in sickle cell anemia, and Prof. Ingo Bechman an expert in neuropathological diseases (Institute of Anatomy, University of Leipzig, Germany). Other IGC researchers involved in this study are Ivo Marguti, Viktória Jeney, Ângelo Chora, Nuno Palha and Sofia Rebelo. This project was funded by Fundação para a Ciência e a Tecnologia (Portugal), GEMI Fund Linde Healthcare and the European Commission's Framework Programme 7.
Related Links / Articles:
Protection from Severe Malaria Explained
Naturally-Occurring Protection Against Severe Malaria Demonstrated
How is it treated? Is there a cure?
Article from National Hear, Lung, and Blood Institute
How Is Sickle Cell Anemia Treated?Sickle cell anemia has no widely available cure. However, treatments can help relieve symptoms and treat complications. The goals of treating sickle cell anemia are to relieve pain; prevent infections, organ damage, and strokes; and control complications (if they occur).
Blood and marrow stem cell transplants may offer a cure for a small number of people who have sickle cell anemia. Researchers continue to look for new treatments for the disease.
Infants who have been diagnosed with sickle cell anemia through newborn screening are treated with antibiotics to prevent infections and receive needed vaccinations. Their parents are educated about the disease and how to manage it. These initial treatment steps have greatly improved the outcome for children who have sickle cell anemia.
Specialists InvolvedPeople who have sickle cell anemia need regular medical care. Some doctors and clinics specialize in treating people who have the disease. Hematologists specialize in treating adults and children who have blood diseases or disorders.
Treating PainMedicines and FluidsMild pain often is treated at home with over-the-counter pain medicines, heating pads, rest, and plenty of fluids. More severe pain may need to be treated in a day clinic, emergency room, or hospital.
The usual treatments for acute (rapid-onset) pain are fluids, medicines, andoxygen therapy (if the oxygen level is low). Fluids help prevent dehydration, a condition in which your body doesn't have enough fluids. Fluids are given either by mouth or through a vein. Your doctor may prescribe antibiotics if you have an infection.
Treatment for mild-to-moderate pain usually begins with acetaminophen (Tylenol®) or nonsteroidal anti-inflammatory drugs (NSAIDs), such as ibuprofen.
If pain continues or becomes severe, stronger medicines called opioids might be needed. Talk with your doctor about the possible benefits and risks of taking strong pain medicine, especially if the medicine will be used for a long period.
HydroxyureaSevere sickle cell anemia can be treated with a medicine called hydroxyurea (hi-DROK-se-yu-RE-ah). This medicine prompts your body to make fetal hemoglobin. Fetal hemoglobin, or hemoglobin F, is the type of hemoglobin that newborns have.
In people who have sickle cell anemia, fetal hemoglobin helps prevent red blood cells from sickling and improves anemia.
Taken daily by mouth, hydroxyurea reduces how often painful sickle cell crises and acute chest syndrome occur. Many people taking hydroxyurea also need fewerblood transfusions and have fewer hospital visits.
Doctors are studying the long-term effects of hydroxyurea on people who have sickle cell anemia. Studies in very young children have shown that hydroxyurea can be given safely and that it improves anemia and hemoglobin F levels while reducing complications of sickle cell anemia. Long-term followup studies of adults treated with hydroxyurea suggest that those treated with the drug survive longer than those not treated with the drug.
Hydroxyurea can reduce the number of white blood cells in your blood, which can raise your risk for infections.
People who take hydroxyurea must have careful medical followup, including blood tests. The dose of this medicine might need to be adjusted to reduce the risk of side effects.
A doctor who has knowledge about hydroxyurea can tell you about the risks and benefits of taking this medicine.
Preventing ComplicationsBlood transfusions are commonly used to treat worsening anemia and sickle cell complications. A sudden worsening of anemia due to an infection or enlarged spleen is a common reason for a blood transfusion.
Some, but not all, people who have sickle cell anemia need regular blood transfusions to prevent life-threatening problems, such as stroke, spleen problems, or acute chest syndrome.
Having routine blood transfusions can cause side effects. Examples include allergic reactions and a dangerous buildup of iron in the body (which must be treated). In general, the blood supply is fairly safe from infections such as hepatitis and HIV.
For more information, go to the Health Topics Blood Transfusion article.
InfectionsInfections can be a major complication of sickle cell anemia throughout life, but especially during childhood. Often, infections can be prevented or treated.
To prevent infections in babies and young children, treatments include:
Adults who have sickle cell anemia also should have flu shots every year and get vaccinated against pneumonia.
Eye DamageSickle cell anemia can damage the blood vessels in the eyes and the retinas. The retinas are the thin layers of tissue at the back of the eyes. Regular checkups with an eye doctor who specializes in diseases of the retina can help detect eye damage.
StrokesStroke prevention and treatment are now possible for children who have sickle cell anemia. Starting at age 2, children who have sickle cell anemia should have routine ultrasound scans of the head. This is called transcranial Doppler (TCD) ultrasound. These scans are used to check the speed of blood flow to the brain.
TCD scans allow doctors to find out which children are at high risk of stroke. Doctors can treat these children with routine blood transfusions to reduce the risk of stroke.
A doctor who has knowledge about blood transfusions and sickle cell disease can tell you about the benefits and risks of this treatment.
Treating Other ComplicationsAcute chest syndrome is a severe and life-threatening complication of sickle cell anemia. If acute (sudden) failure of the liver and kidneys also occurs, it's called acute multiple organ failure.
Treatment for these complications usually occurs in a hospital and may include oxygen therapy, blood transfusions, antibiotics, pain medicine, and balancing body fluids.
Leg ulcers (sores) due to sickle cell anemia can be very painful. Ulcers can be treated with cleansing solutions and medicated creams or ointments.
Skin grafts might be needed if the leg ulcers are ongoing. Bed rest and keeping the legs raised to reduce swelling are helpful. If you have a lot of pain from leg ulcers, your doctor may recommend a strong pain medicine.
Your doctor might recommend gallbladder surgery if the presence of gallstones leads to gallbladder disease.
Priapism (a painful erection in males) can be treated with fluids, medicines, or surgery.
Regular Health Care for ChildrenChildren who have sickle cell anemia need routine health care (just like children who don't have the disease). They need to have their growth checked regularly. They also need to get the routine shots that all children get.
All children younger than 2 years old should see their doctors often. Children who have sickle cell anemia may need even more checkups. After age 2, children who have sickle cell anemia may not need to see their doctors as often, but they usually still need checkups at least every 6 months.
These visits are a time for parents to talk with their child's doctor and ask questions about the child's care. Talk with your child's doctor about eye checkups and whether your child needs an ultrasound scan of the brain.
Until age 5, daily penicillin is given to most children who have sickle cell anemia. Doctors also give many children a vitamin called folic acid (folate) to help boost red blood cell production.
Young children who have sickle cell anemia should have regular checkups with a hematologist (a blood specialist).
New TreatmentsResearch on blood and marrow stem cell transplants, gene therapy, and new medicines for sickle cell anemia is ongoing. The hope is that these studies will provide better treatments for the disease. Researchers also are looking for a way to predict the severity of the disease.
Blood and Marrow Stem Cell TransplantA blood and marrow stem cell transplant can work well for treating sickle cell anemia. This treatment may even offer a cure for a small number of people.
The stem cells used for a transplant must come from a closely matched donor. The donor usually is a close family member who doesn't have sickle cell anemia. This limits the number of people who may have a donor.
The transplant process is risky and can lead to serious side effects or even death. However, new transplant approaches may improve treatment for people who have sickle cell anemia and involve less risk.
Blood and marrow stem cell transplants usually are used for young patients who have severe sickle cell anemia. However, the decision to give this treatment is made on a case-by-case basis.
Researchers continue to look for sources of bone marrow stem cells—for example, blood from babies' umbilical cords. They also continue to look for ways to reduce the risks of this procedure.
For more information, go to the Health Topics Blood and Marrow Stem Cell Transplant article.
Gene TherapyGene therapy is being studied as a possible treatment for sickle cell anemia. Researchers want to know whether a normal gene can be put into the bone marrow stem cells of a person who has sickle cell anemia. This would cause the body to make normal red blood cells.
Researchers also are studying whether they can "turn off" the sickle hemoglobin gene or "turn on" a gene that makes red blood cells behave normally.
New MedicinesResearchers are studying several medicines for sickle cell anemia. They include:
You can’t prevent sickle cell anemia, because it’s an inherited disease. If a person is born with it, steps should be taken to reduce complications. (For more information, go to "Living With Sickle Cell Anemia.")
People who are at high risk of having a child with sickle cell anemia and are planning to have children may want to consider genetic counseling. A counselor can explain the risk (likelihood) of having a child who has the disease. He or she also can help explain the choices that are available.
You can find information about genetic counseling from health departments, neighborhood health centers, medical centers, and clinics that care for people who have sickle cell anemia.
Article from National Hear, Lung, and Blood Institute
How Is Sickle Cell Anemia Treated?Sickle cell anemia has no widely available cure. However, treatments can help relieve symptoms and treat complications. The goals of treating sickle cell anemia are to relieve pain; prevent infections, organ damage, and strokes; and control complications (if they occur).
Blood and marrow stem cell transplants may offer a cure for a small number of people who have sickle cell anemia. Researchers continue to look for new treatments for the disease.
Infants who have been diagnosed with sickle cell anemia through newborn screening are treated with antibiotics to prevent infections and receive needed vaccinations. Their parents are educated about the disease and how to manage it. These initial treatment steps have greatly improved the outcome for children who have sickle cell anemia.
Specialists InvolvedPeople who have sickle cell anemia need regular medical care. Some doctors and clinics specialize in treating people who have the disease. Hematologists specialize in treating adults and children who have blood diseases or disorders.
Treating PainMedicines and FluidsMild pain often is treated at home with over-the-counter pain medicines, heating pads, rest, and plenty of fluids. More severe pain may need to be treated in a day clinic, emergency room, or hospital.
The usual treatments for acute (rapid-onset) pain are fluids, medicines, andoxygen therapy (if the oxygen level is low). Fluids help prevent dehydration, a condition in which your body doesn't have enough fluids. Fluids are given either by mouth or through a vein. Your doctor may prescribe antibiotics if you have an infection.
Treatment for mild-to-moderate pain usually begins with acetaminophen (Tylenol®) or nonsteroidal anti-inflammatory drugs (NSAIDs), such as ibuprofen.
If pain continues or becomes severe, stronger medicines called opioids might be needed. Talk with your doctor about the possible benefits and risks of taking strong pain medicine, especially if the medicine will be used for a long period.
HydroxyureaSevere sickle cell anemia can be treated with a medicine called hydroxyurea (hi-DROK-se-yu-RE-ah). This medicine prompts your body to make fetal hemoglobin. Fetal hemoglobin, or hemoglobin F, is the type of hemoglobin that newborns have.
In people who have sickle cell anemia, fetal hemoglobin helps prevent red blood cells from sickling and improves anemia.
Taken daily by mouth, hydroxyurea reduces how often painful sickle cell crises and acute chest syndrome occur. Many people taking hydroxyurea also need fewerblood transfusions and have fewer hospital visits.
Doctors are studying the long-term effects of hydroxyurea on people who have sickle cell anemia. Studies in very young children have shown that hydroxyurea can be given safely and that it improves anemia and hemoglobin F levels while reducing complications of sickle cell anemia. Long-term followup studies of adults treated with hydroxyurea suggest that those treated with the drug survive longer than those not treated with the drug.
Hydroxyurea can reduce the number of white blood cells in your blood, which can raise your risk for infections.
People who take hydroxyurea must have careful medical followup, including blood tests. The dose of this medicine might need to be adjusted to reduce the risk of side effects.
A doctor who has knowledge about hydroxyurea can tell you about the risks and benefits of taking this medicine.
Preventing ComplicationsBlood transfusions are commonly used to treat worsening anemia and sickle cell complications. A sudden worsening of anemia due to an infection or enlarged spleen is a common reason for a blood transfusion.
Some, but not all, people who have sickle cell anemia need regular blood transfusions to prevent life-threatening problems, such as stroke, spleen problems, or acute chest syndrome.
Having routine blood transfusions can cause side effects. Examples include allergic reactions and a dangerous buildup of iron in the body (which must be treated). In general, the blood supply is fairly safe from infections such as hepatitis and HIV.
For more information, go to the Health Topics Blood Transfusion article.
InfectionsInfections can be a major complication of sickle cell anemia throughout life, but especially during childhood. Often, infections can be prevented or treated.
To prevent infections in babies and young children, treatments include:
- Daily doses of antibiotics. Treatment may begin as early as 2 months of age and continue until the child is at least 5 years old.
- All routine vaccinations (including a yearly flu shot), plus the pneumococcal vaccine.
Adults who have sickle cell anemia also should have flu shots every year and get vaccinated against pneumonia.
Eye DamageSickle cell anemia can damage the blood vessels in the eyes and the retinas. The retinas are the thin layers of tissue at the back of the eyes. Regular checkups with an eye doctor who specializes in diseases of the retina can help detect eye damage.
StrokesStroke prevention and treatment are now possible for children who have sickle cell anemia. Starting at age 2, children who have sickle cell anemia should have routine ultrasound scans of the head. This is called transcranial Doppler (TCD) ultrasound. These scans are used to check the speed of blood flow to the brain.
TCD scans allow doctors to find out which children are at high risk of stroke. Doctors can treat these children with routine blood transfusions to reduce the risk of stroke.
A doctor who has knowledge about blood transfusions and sickle cell disease can tell you about the benefits and risks of this treatment.
Treating Other ComplicationsAcute chest syndrome is a severe and life-threatening complication of sickle cell anemia. If acute (sudden) failure of the liver and kidneys also occurs, it's called acute multiple organ failure.
Treatment for these complications usually occurs in a hospital and may include oxygen therapy, blood transfusions, antibiotics, pain medicine, and balancing body fluids.
Leg ulcers (sores) due to sickle cell anemia can be very painful. Ulcers can be treated with cleansing solutions and medicated creams or ointments.
Skin grafts might be needed if the leg ulcers are ongoing. Bed rest and keeping the legs raised to reduce swelling are helpful. If you have a lot of pain from leg ulcers, your doctor may recommend a strong pain medicine.
Your doctor might recommend gallbladder surgery if the presence of gallstones leads to gallbladder disease.
Priapism (a painful erection in males) can be treated with fluids, medicines, or surgery.
Regular Health Care for ChildrenChildren who have sickle cell anemia need routine health care (just like children who don't have the disease). They need to have their growth checked regularly. They also need to get the routine shots that all children get.
All children younger than 2 years old should see their doctors often. Children who have sickle cell anemia may need even more checkups. After age 2, children who have sickle cell anemia may not need to see their doctors as often, but they usually still need checkups at least every 6 months.
These visits are a time for parents to talk with their child's doctor and ask questions about the child's care. Talk with your child's doctor about eye checkups and whether your child needs an ultrasound scan of the brain.
Until age 5, daily penicillin is given to most children who have sickle cell anemia. Doctors also give many children a vitamin called folic acid (folate) to help boost red blood cell production.
Young children who have sickle cell anemia should have regular checkups with a hematologist (a blood specialist).
New TreatmentsResearch on blood and marrow stem cell transplants, gene therapy, and new medicines for sickle cell anemia is ongoing. The hope is that these studies will provide better treatments for the disease. Researchers also are looking for a way to predict the severity of the disease.
Blood and Marrow Stem Cell TransplantA blood and marrow stem cell transplant can work well for treating sickle cell anemia. This treatment may even offer a cure for a small number of people.
The stem cells used for a transplant must come from a closely matched donor. The donor usually is a close family member who doesn't have sickle cell anemia. This limits the number of people who may have a donor.
The transplant process is risky and can lead to serious side effects or even death. However, new transplant approaches may improve treatment for people who have sickle cell anemia and involve less risk.
Blood and marrow stem cell transplants usually are used for young patients who have severe sickle cell anemia. However, the decision to give this treatment is made on a case-by-case basis.
Researchers continue to look for sources of bone marrow stem cells—for example, blood from babies' umbilical cords. They also continue to look for ways to reduce the risks of this procedure.
For more information, go to the Health Topics Blood and Marrow Stem Cell Transplant article.
Gene TherapyGene therapy is being studied as a possible treatment for sickle cell anemia. Researchers want to know whether a normal gene can be put into the bone marrow stem cells of a person who has sickle cell anemia. This would cause the body to make normal red blood cells.
Researchers also are studying whether they can "turn off" the sickle hemoglobin gene or "turn on" a gene that makes red blood cells behave normally.
New MedicinesResearchers are studying several medicines for sickle cell anemia. They include:
- Decitabine. Like hydroxyurea, this medicine prompts the body to make fetal hemoglobin. Fetal hemoglobin helps prevent red blood cells from sickling and improves anemia. Decitabine might be used instead of hydroxyurea or added to hydroxyurea.
- Adenosine A2a receptor agonists. These medicines may reduce pain-related complications in people who have sickle cell anemia.
- 5-HMF. This natural compound binds to red blood cells and increases their oxygen. This helps prevent the red blood cells from sickling.
You can’t prevent sickle cell anemia, because it’s an inherited disease. If a person is born with it, steps should be taken to reduce complications. (For more information, go to "Living With Sickle Cell Anemia.")
People who are at high risk of having a child with sickle cell anemia and are planning to have children may want to consider genetic counseling. A counselor can explain the risk (likelihood) of having a child who has the disease. He or she also can help explain the choices that are available.
You can find information about genetic counseling from health departments, neighborhood health centers, medical centers, and clinics that care for people who have sickle cell anemia.
Alternative Ideas: (see also Integrative / Holistic Research page)
Sickle Cell is treated primarily with pain killers, hydration, Hydroxyurea, antibiotics, immunizations, blood transfusions, stroke assessments, supplemental oxygen, stem cell transplant, temperature control, and time. Sickle Cell is best managed in a preventative way by using various methods: Hydroxyurea, keeping hydrated, keeping warm, lots of rest, healthy food, health lifestyle, low stress, and regular doctor check ups.
There are experimental methods being tested such as: gene therapy, Nitric oxide, and drugs to boost fetal hemoglobin production.
There are integrative and alternative methods that are also being explored: thiocyanate, prickly ash, and infrared heat and heating pads. Every form of healing has a place. It is important to explore all areas of healing, both allopathic / traditional medicine and integrative / holistic methods. What may work for some may not work for others. It is important to understand that integrative / holistic approaches may take longer to see effects, but can be worth it in the long run as you may avoid unwanted long term side effects of traditional / allopathic approaches. Integrative / holistic approaches can have side effects too, so it is important to speak with a specialist (herbalist, pharmacist, naturopath, osteopath, homeopath, etc) and keep your doctor informed on what you are taking as some remedies may interact with other drugs you may be taking. Holistic approaches may be a nice alternative to the harsh and addictive drugs that are prescribed for pain. It may be possible to explore integrative approaches while you are being treated traditionally, but consult your physicians (GP and hematologists, etc) first. If your condition is severe see your doctor right away.
Sickle Cell is treated primarily with pain killers, hydration, Hydroxyurea, antibiotics, immunizations, blood transfusions, stroke assessments, supplemental oxygen, stem cell transplant, temperature control, and time. Sickle Cell is best managed in a preventative way by using various methods: Hydroxyurea, keeping hydrated, keeping warm, lots of rest, healthy food, health lifestyle, low stress, and regular doctor check ups.
There are experimental methods being tested such as: gene therapy, Nitric oxide, and drugs to boost fetal hemoglobin production.
There are integrative and alternative methods that are also being explored: thiocyanate, prickly ash, and infrared heat and heating pads. Every form of healing has a place. It is important to explore all areas of healing, both allopathic / traditional medicine and integrative / holistic methods. What may work for some may not work for others. It is important to understand that integrative / holistic approaches may take longer to see effects, but can be worth it in the long run as you may avoid unwanted long term side effects of traditional / allopathic approaches. Integrative / holistic approaches can have side effects too, so it is important to speak with a specialist (herbalist, pharmacist, naturopath, osteopath, homeopath, etc) and keep your doctor informed on what you are taking as some remedies may interact with other drugs you may be taking. Holistic approaches may be a nice alternative to the harsh and addictive drugs that are prescribed for pain. It may be possible to explore integrative approaches while you are being treated traditionally, but consult your physicians (GP and hematologists, etc) first. If your condition is severe see your doctor right away.
- Please consult your doctor no matter what method you choose as there can be complications, contraindications, allergic reactions, and side effects to consider with all methods of healing. And it is always wise to get a second opinion from other doctors just in case.
University of Illinois - Hospital and Health Sciences System
Chicago Woman Cured of Sickle Cell Disease
http://hospital.uillinois.edu/Patient_Care_Services/Sickle_Cell/Managing_Sickle_Cell.html
National Heart, Lung, and Blood Institute
http://www.nhlbi.nih.gov/health/health-topics/topics/sca/treatment.html
Mayo Clinic - Treatment
http://www.mayoclinic.com/health/sickle-cell-anemia/DS00324/DSECTION=treatments-and-drugs
University of Maryland Medical Center
http://umm.edu/health/medical/reports/articles/sickle-cell-disease
University of Illinois - Hospital and Health Sciences System
http://hospital.uillinois.edu/Patient_Care_Services/Sickle_Cell/Managing_Sickle_Cell.html
Medscape
http://emedicine.medscape.com/article/205926-treatment
The New York times - Health Guide
http://www.nytimes.com/health/guides/disease/sickle-cell-anemia/
- Disclaimer:
Information or materials on the website are not intended to treat or replace your doctor’s advice. It is highly recommended that patients contact their haematologists or care provider for adequate care and treatment